 2013, Vol. 18
2013, Vol. 18



2 中国科学院大气物理研究所大气边界层物理和大气化学国家重点实验室, 北京 100029
2 State Key Laboratory of Atmospheric Boundary Layer Physics and Atmospheric Chemistry, Institute of Atmospheric Physics, Chinese Academy of Sciences, Beijing 100029
1 引言
反硝化过程使氮素从硝态氮(NO3–)转化为氮气(N2),这是维系闭合全球氮循环所必需的氮素形态转化环节,土壤微生物反硝化过程是该环节的重要组成。在土壤微生物反硝化过程中,微生物(包括细菌和真菌)利用碳底物的氧化提供电子,将硝态氮逐步还原成一氧化氮(NO)、氧化亚氮(N2O)和N2。N2本身不具有化学和生物活性,而大气 N2O却是一种重要的温室气体,并可参与破坏平 流层臭氧,大气NO是臭氧(O3)形成的前提 物,也是形成酸雨的主要物质,因而它们的土壤排放,尤其是人为活动影响(如施氮肥等)下的土壤排放很受关注(IPCC,2007)。初始硝态氮向这3种气体的转化率总和,定义为总反硝化速率。对反硝化速率及其各气体产物所占比例的直接测定是研究氮循环过程机理和认识N2O和NO排放规律的基础,但却又是一个难题,这主要是因为大气背景中的高N2浓度(78%)使反硝化N2排放速率的直接测定极其困难(Davidson and Seitzinger, 2006;Groffman et al., 2006)。
目前测定土壤反硝化速率的方法主要有:(1)乙炔(C2H2)抑制法(Yoshinari and Knowles, 1976);(2)15N示踪法(Nömmik,1956);(3)直接测定N2法(Swerts et al., 1995;Scholefield et al., 1997a;Butterbach-Bahl et al., 2002)。
乙炔抑制法因其操作简便并可适用于大田而得到了广泛的应用,但它也有不可忽视的缺点:(1)高浓度C2H2可以同时抑制硝化和反硝化作用(Klemedtsson et al., 1990),造成在有氧条件下对反硝化速率的低估;(2)C2H2难以均匀扩散到被检测的整个土体中,抑制效果欠佳(Jordan et al., 1998);(3)土壤NO3–浓度较低时,C2H2对N2O还原酶抑制不完全(Simarmata et al., 1993);(4)当氧气(O2)和较高浓度C2H2存在时,NO会被C2H2催化氧化成二氧化氮,造成对反硝化速率的低估(BollmannConrad et al., 1997);(5)可测定总反硝化速率,但不能测定单个反硝化产物速率。
15N同位素示踪法需要向土壤中引入标记物,此过程会对土壤造成较大扰动,也会刺激微生物氮转化过程(Butterbach-Bahl et al., 2002),从而给测定结果造成较大误差。更大的问题是,该方法检测N2的灵敏度不高,只能检测>500 gN2–N ha–1 d–1 [或>50 mgN2–N h–1 kg–1干土(d.s.)]的排放速率(Butterbach-Bahl et al., 2002)。此外,该方法测定过程比较复杂、成本也较高(Butterbach-Bahl et al., 2002;Groffman et al., 2006)。
直接测定N2法由Stefanson and Greenlan(1970)发明,后经Swerts et al.(1995)、Scholefield et al.(1997a)、Butterbach-Bahl et al.(2002)、Cárdenas et al.(2003)和Molstad et al.(2007)等人的改进而进一步发展。该方法的基本原理是,用高纯氦(He)或He-O2混合气体[O2可设定为≤20%(自由大气浓度)]置换去除土壤孔隙和密闭培养容器气室中高N2含量的空气,从而实现对土壤排放的N2的直接测定。和C2H2与15N同位素示踪法相比,直接测定N2法具有如下优点:(1)不用向土壤中引入标记物,这对于研究半自然和自然环境条件下的N2排放非常重要;(2)不用添加C2H2抑制剂,避免了C2H2可能引起的不良影响;(3)具有比15N示踪法高得多的检测灵敏度(2.4 gN2-N ha–1 d–1或0.3 μgN2-N h–1 kg–1d.s.)(Butterbach-Bahl et al., 2002)。正因为这些优点,直接测定N2法的发展和应用近来很受关注(Groffman et al., 2006),并根据不同研究目的具体需求不同,国际上发展出了四类略有不同的氦环境培养—气体同步直接测定系统,分别以Swerts et al.(1995)、Scholefield et al.(1997a)、Butterbach-Bahl et al.(2002)以及Molstad et al.(2007)报道的测定系统为代表。对于土壤反硝化N2排放的直接测定,以Butterbach-Bahl et al.(2002)系统最为灵敏。
尽管直接测定N2法得到很大的发展,但目前仍存在一些问题:(1)很难同步检测反硝化过程中多种(中间)产物产生速率,不利于有效地刻画陆地生态系统中反硝化关键过程及其驱动机制;(2)测定数据难以实现培养前后的碳和氮平衡(Scholefield et al., 1997a,1997b;Cárdenas et al., 2003),从而难以做到对反硝化过程进行准确定量。这预示着,当前的直接测定N2法仍需进一步改进。为此,德国卡尔斯鲁厄技术研究所与中国科学院大气物理研究所最近对Butterbach-Bahl et al.(2002)系统做了一些改进,设计了一套新的氦环境培养—气体同步直接测定系统(Wang et al., 2011)。该系统的主要改进是:简化了培养容器的气路设计;改进了置换方式(由吹扫式改为负压式),大大缩短了置换时间;将检测N2的非放射性脉冲放电氦离子化检测器(PDHID)改成了通用型的热导检测器(TCD);实现对反硝化气体(N2、N2O和NO)和CO2排放的同步测定;建立了三阶段培养法(低温有氧置换阶段—低温无氧置换阶段—常温厌氧测定阶段),目的是为了减少气体置换过程对气体排放的影响(Wang et al., 2011)。
为检验该新建测定系统是否可以实现培养前后的氮平衡,并了解其测定土壤反硝化速率和反硝化产物比的准确性,本文用采自华北地区盐碱地的农田土壤,测试了该系统对两个初始硝态氮含量水平的反硝化过程测定效果。
2 材料与方法 2.1 氦环境培养—气体同步直接测定系统简介
该测定系统由培养容器、气路系统和分析检测系统三部分组成(图 1)。培养容器是3个完全相同的不锈钢圆柱形容器(内径13.9~14.5 cm,外径18.9 cm,高5.6 cm),每个容器可放置4个环刀土柱(放入土柱后,培养容器所剩空间约为408 mL,称为气室)。培养容器置于恒温水浴箱(130×73×84 cm3)中,以控制土壤培养温度。培养容器内部空间与系统的其他部分通过不锈钢管(内径2 mm,外径3 mm)路连通。气路系统由气源[高纯He(99.9997%)、高纯O2(99.999%)和N2标气(20 μmol mol–1 N2,He为底气)]、质量流量计、电磁阀、控制模块、真空泵等组成。分析检测系统由带有TCD的气相色谱仪(美国安捷伦科技有限公司出品的Agilent micro-GC 3000,用于测定N2)、带有电子捕获检测器(ECD)和氢火焰离子化检测器(FID)的气相色谱仪(美国惠普公司出品的HP 5890,用于测定N2O和CO2)和化学发光氮氧化物分析仪(美国赛默飞世尔科技公司出品的Thermo 42i,用于测定NO)组成。关于该系统的更详细介绍,见Wang et al.(2011)。

|
图1 氦环境培养—气体同步直接测定系统构造图(P:泵;FC:质量流量计: :电磁阀)
Figure1 Schematic diagram of the gas-flow-soil-core system (P: pump; FC: flow controller; : magnetic valves) :电磁阀)
Figure1 Schematic diagram of the gas-flow-soil-core system (P: pump; FC: flow controller; : magnetic valves)
|
整个系统由数据采集系统计算机程序软件IDAS(IFU Data Acquisition System)控制。系统 运行时,计算机程序软件通过各控制模块发出指令,设置和调节气路流量,执行电磁阀的开关动作,从而改变管线中气体流速和方向,以达到气体置换、平衡和采样分析的目的。置换、平衡和采样分析步骤具体如下:
(1)气体置换。用80%高纯He和20%高纯O2构成的混合气体(以下简称置换气)置换培养容器 内土壤孔隙中和气室内含高浓度N2的空气,直 至其全部为置换气所取代。置换过程采用负压式置换,即首先将培养容器内含有高浓度N2的气体抽出,使其到达负压状态,然后注入置换气以平衡压力。这样的过程在3个培养容器间反复交替进行,直至气体置换完成(如图 1虚线气路所示)。完成置换的时间需要20~40 h不等,依土壤质地、含水量而异。置换气的构成和置换温度等条件由试验目的确定。气体置换时间的确定方法:首先将供试土壤进行灭菌,灭菌后的土壤放入培养容器内,用置换气去置换培养容器内的高浓度N2的空气,每置换土壤空气一段时间(3~5 h),测定一次培养容器顶部空间N2浓度的变化,直到测定的顶部空间N2浓度的变化恒定时,认为气体置换过程完成,而这个恒定的N2浓度变化率即为外界N2的渗漏率,而这期间每次气体置换时间之和即为该类型土壤在该湿度下的气体置换时间。
(2)建立气体流动平衡。为模拟野外自然条件下土壤表层空气不断流动的状态,同时也为避免气室内气体的过度累积对测定结果的影响,在气体置换结束后的排放速率测定间隙,需要使土柱上方气室始终处于气体流动的平衡状态。用N2浓度为20 μmol mol–1的标气(底气为高纯He)以20 mL min–1的流速吹扫培养容器的气室,以在土壤内部建立自然的气体浓度梯度,同时也平衡气室的压力(如 图 1实线气路所示)。在所有气体排放速率测定间隙,培养容器中始终保持这种气体流动平衡状态。
(3)气体采样分析。气体置换结束后,以8~24 h的时间间隔测定培养容器气室中气体(N2、N2O、NO和CO2)的排放速率,直到氮素气体排放速率降低到检测限附近为止。每次排放速率测定均分别以45和15 min间隔测定5次N2和其他气体(N2O、NO和CO2)的浓度,根据5次浓度的变化率计算N2、N2O、NO和CO2 的排放速率。
(4)系统渗漏率测定。在空的培养容器内进行,不放置供试土壤样品,采用高纯He置换培养容器中的空气,使其全部为He所取代,然后以一定时间间隔测定培养容器空间的气体浓度变化,即为系统渗漏率,该方法可作为检验系统气密性的日常测定方法。
2.2 供试土壤供试土壤采自山西省永济市董村农场(34°56'N,110°43'E)典型冬小麦—夏玉米轮作农田(李明等,2009),供试土壤的理化性质见表 1。采回的鲜土经风干至质量含水量为18%左右,过2 mm筛、除杂、混匀,置于4 ℃冷藏箱内干燥保存。每次试验开始前2周,加水(去离子水)调节土壤湿度为25%(质量含水量,约44%土壤充水孔隙度,缩写WFPS),将土壤在低温条件下(4 ℃)进行预培养,预培养期间,每天加水保持土壤湿度25%,并且每天开冷藏箱门通气2~3次,以防止培养箱内供试土壤因O2缺乏而发生反硝化作用消耗底物。
| 表 1 供试土壤理化性质 Table 1 Physical and chemical properties of soil samples |

本试验设置两个初始NO3--N浓度水平[用硝酸钾(KNO3)调节],分别为大约10和100 mgN kg–1d.s.(以下简称10N和100N)。每个NO3--N水平的土壤初始可溶性有机碳(DOC)含量均维持在大约300 mgC kg–1d.s.(用葡萄糖调节)。在完全厌氧条件下培养和测定反硝化气体(N2、N2O和NO)和CO2 排放速率动态,以研究初始NO3--N含量变化对25 ℃条件下土壤反硝化气体(N2、N2O和NO)和CO2排放的影响。
对于每个NO3--N浓度水平处理,用不锈钢环刀(内径5.6 cm,外径6 cm,高4 cm)取12个土柱,每个土柱湿重130 g 左右,随机分成3组,分别置于3个培养容器内,每个培养容器放置4个环刀土柱。在取环刀土柱之前,先取土壤样品测定其中的铵态氮(NH4+-N)、NO3--N和DOC含量,作为调节培养土柱初始矿质氮和DOC含量的参考依据。在取环刀土柱的同时,再取土壤样品测定其中的NH4+-N、NO3--N、DOC、微生物碳(SMBC)、微生物氮(SMBN)含量和含水量,作为准确确定培养土柱的初始碳、氮、水含量的依据。在密闭培养容器以进行气体置换之前,向每个环刀土柱中均匀添加5 mL KNO3和葡萄糖的混合溶液,使其达到土壤初始NO3--N和DOC含量设定值。加入溶液后,土壤湿度由25%增加至30%(约53% WFPS)。气体排放速率测定结束后,立即测定土壤样品中的NH4+-N、NO3--N、DOC、SMBC、SMBN和含水量,以定量这些变量在培养测定过程中的变化。
2.4 培养方法
本研究采用低温置换,即在整个置换过程中使土柱温度维持在2 ℃左右。低温条件可以避免置换过程发生氧化反应而消耗初始NH4+和DOC等。在低温条件下首先采用上述He-O2混合气体进行置换,以防止置换过程中发生反硝化作用而消耗底物NO3-。为了测定反硝化速率,在低温置换后期,需将O2源切断,使土柱内形成完全厌氧状态。置换结束后,将土壤温度迅速调节至25 ℃,进行反硝化气体和CO2排放速率动态的测定。培养容器中环刀土柱的上述培养过程可用图 2所示的三阶段表示,分别是低温有氧置换阶段(P1)、低温厌氧置换阶段(P2)和常温厌氧培养测定阶段(P3)。

|
图2 培养期间各阶段的条件设置和测定排放速率的气体种类 Figure2 Setting conditions and gas species to measure in each incubation period |
在P1阶段,用80% He和20% O2构成的混合气体,以250 mL min–1的流速每置换土壤空气一段时间(先后分别为5、10、8 h),测定一次N2O、NO和CO2的排放速率。测定时首先将置换停止,并平衡30 min 左右,然后进行测定,测定时间为60 min,测定期间每间隔15 min测定一次气室内的N2O、NO和CO2浓度,共测定5次用于排放速率的计算。P1阶段有氧置换(大约30 h)结束后至P2阶段厌氧置换前,先后测定两次低温有氧条件下的N2 排放速率,测定时间为3 h,测定期间每间隔45 min测定一次气室内的N2浓度,共测定5次用于排放速率的计算。
在P2阶段,继续保持低温,但切断了O2源,进行两次纯He置换(置换气体流速为200 mL min–1),每次置换2 h。每次置换结束后都测定一次四种气体的排放速率,每次测定之前平衡大约30 min。之后继续保持低温,持续间歇性地测定4次4种气体的排放速率,每次排放速率测定持续5 h左右,每两次测定之间平衡2 h左右。
在P3阶段,首先在流动气体平衡条件下将水浴快速升温至25 ℃,然后每间隔8 h 测定一次4种气体的排放速率,到气体排放高峰过去后降为每间隔12 h或24 h测定一次,直至各氮素气体排放速率接近检测限为止。在所有气体排放速率测定间隙,培养容器顶部空间始终有20 mL min–1的平衡气流过。
所有样品的测定均于采样后1 h内完成。
气室内气体的N2浓度由安捷伦micro GC 3000微型气相色谱在线采样分析。对于每个气体样品测定,微型气相色谱的内置真空泵以2 mL min–1的流速抽取吹扫土柱上方气室流出的气体,使其连续流过微型气相色谱的气体样品定量管20 s,然后进样分析,用TCD检测器检测N2信号,并用几乎同步分析的N2标气(20 μmol mol–1,底气为高纯He)进行浓度标定。微型气相色谱的配置条件如下:分子筛色谱柱(14 m×320 μm×12 μm);柱温80 ℃;检测器温度50 ℃;进样口温度80 ℃;载气高纯He(99.9997%,流速20 mL min–1);出峰时间1.8 min。
分析N2O、NO和CO2的样气从系统采气口用60 mL注射器以20 mL min–1的速度(与采样时系统的进气速度一致)采样3 min,所采集的60 mL样气,20 mL用于测定N2O和CO2(视浓度高低决定是否需要稀释),余下40 mL注入充有1.8 L高纯N2(99.999%)的气袋(大连普莱特气体包装有限公司出品),用于测定NO。检测N2O和CO2的色谱配置条件如下:载气高纯N2,流速30 mL min–1;检测器ECD温度350 ℃,FID温度220 ℃;色谱柱PorapakQ,柱温55 ℃;导入ECD的高CO2缓冲气体(CO2/N2=10/90,流速约3 mL min–1)。详细的色谱配置条件请参看Zheng et al.(2008)和Wang et al.(2011)。
N2O采用外标工作曲线来标定。CO2和N2采用单点标定,其标气浓度分别为354 μmol mol–1 CO2(高纯N2为底气)和20 μmol mol–1 N2(高纯He为底气)。测定NO的氮氧化物分析仪每次试验之前标定一次。所有标准气体均由北京氦普北分气体工业有限公司提供。
测定系统检测N2、N2O、NO和CO2浓度的分析精度分别为0.2、0.005、0.045和1.3 μmol mol–1。
2.6 排放速率计算
N2标气(20 μmol mol–1 N2)以一定流速(20 mL min–1)吹扫土柱顶部气室,并携带培养容器内的样气通过尾气放空口流出,这种采样方式对气室内的气体造成了稀释,使测得的流出气体N2、N2O、NO和CO2浓度不能直接代表土壤排放气体的真实 浓度。因此,在用于计算排放速率之前需对其进行矫正。矫正公式(Butterbach-Bahl et al., 2002)如下
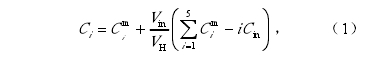
采用矫正后的气体浓度值Ci和以下公式(Wang et al., 2011)可计算上述气体的排放速率:
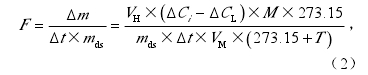
由上述气体浓度检测精度、浓度测定时间间隔、培养容器气室体积及测定土壤样品干重,可得N2、N2O、NO和CO2的排放速率检测限依次为0.23、0.02、0.08 µgN h–1 kg–1d.s.和1.9 µgC h–1 kg–1 d.s.。
2.7 土壤辅助参数测定
土壤质量含水量(SWC)的测定采用烘干称重法;NH4+-N的测定采用2 M KCl浸提(水土比5:1)—靛酚蓝比色法;NO3--N和DOC的测定采用无CO2去离子水浸提(水土比5:1)浸提液用0.45 μm聚醚砜膜过滤,用离子色谱仪(瑞士万通中国有限公司出品的790通用型)测定NO3--N,用碳氮分析仪(德国耶拿公司出品的Multi N/C 3000型)测定DOC(引自《土壤农化分析第三版》)(鲍士旦,2000)。SMBC和SMBN的测定采用氯仿熏蒸法(Sparling and West, 1988),用碳氮分析仪测定浸提液的总有机碳(TOC)和总氮(TN)含量,并用熏蒸和未熏蒸土壤样品的TOC和TN之差分别表示SMBC和SMBN含量。
2.8 统计分析方法
统计检验采用Excel数据分析工具中的双样本等方差假设分析,统计显著水平由t检验给出。
3 结果 3.1 反硝化气体(N2、N2O和NO)与CO2的排放速率动态10N和100N两处理反硝化气体(N2、N2O和NO)和CO2的排放动态如图 3所示。

|
图3 反硝化气体(N 2 、N 2 O、NO)和 CO 2 的排放动态:(a) 10N 处理;(b) 100N 处理 Figure2 SEmission dynamics of denitrification gases (N 2 , N 2 O, and NO) and CO 2 from incubated soils: (a) 10N; (b) 100N |
P1阶段:随着温度逐渐降低,土壤呼吸作用减弱,两处理CO2的排放速率均逐渐降低至100(10N) ~130(100N)μgC h–1 kg–1d.s.。两处理的初始CO2排放速率高是因为土壤样品的温度一开始高于此阶段的设定值(2 ℃左右),后来逐渐降低并稳定是因为土壤温度逐渐降低到设定值,呼吸作用也逐渐减弱,并达到稳定状态。置换气中的高O2含量抑制了反硝化作用,低温抑制了硝化作用,因而此阶段测得的N2O和NO排放速率都很低,接近系统检测限。此阶段后期测得的10N处理的N2排放速率为0.5 μgN h–1 kg–1d.s.,100N为0.8 μgN h–1 kg–1 d.s.,100N显著高于10N(统计检验显著水平p<0.05),且两处理N2排放速率均显著高于N2O和NO排放速率之和(p<0.01)。
P2阶段:置换气切换为纯He后,虽然培养温度仍然为2 ℃左右,但反硝化作用还是因缺O2而明显增强,致使N2、N2O和NO排放速率均比P1阶段明显增加(p<0.01)。至该阶段后期,10N处理的N2、N2O和NO排放速率分别增加到37、6和6 μgN h–1 kg–1d.s.,100N处理分别增加到61、26和17 μgN h–1 kg–1d.s.,后一处理3种气体的排放速率依次比前一处理高大约1倍(p<0.01)、4倍(p<0.01)和3倍(p<0.01)。尽管此阶段的置换气无O2,但两处理的CO2排放速率较上一阶段并未受显著影响,始终维持在120(10N)~170(100N)μgC h–1 kg–1d.s.左右。该阶段CO2排放来自于微生物无氧呼吸,而P1阶段的CO2排放来自于微生物有氧呼吸,尽管P2阶段CO2排放速率较P1阶段末的CO2排放无显著差异,但CO2其实来自于不同的微生物呼吸过程。
P3阶段:在无O2条件下将培养温度升至25 ℃后,反硝化作用剧烈发生,N2、N2O、NO和CO2排放速率均比P2阶段迅速增加,此阶段4种气体的第一次测定值分别比上一阶段最后一次测定值高300~552 μgN h–1 kg–1d.s.(p<0.01)、143~492 μgN h–1 kg–1d.s.(p<0.01)、60~195 μgN h–1 kg–1d.s.(p<0.01)和495~855 μgC h–1 kg–1d.s.(p< 0.01),除N2的排放速率增加幅度是10N大于100N外,其他3种气体均是100N大于10N处理。10N处理的N2O和NO排放速率在升温后2 h左右同时达到最大值(分别为149±5和67±5 μgN h–1 kg–1 d.s.),100N处理的N2O和NO排放速率在升温后 9 h左右才同时达到最大值(分别为637±20和412±11 μgN h–1 kg–1d.s.),并且分别比10N最 大值高3倍(p<0.01)和5倍(p<0.01)。10N处理的N2排放速率峰值(590±27 μgN h–1 kg–1 d.s.)与N2O和NO几乎同步,但分别比N2O和NO排放速率峰值高4倍(p<0.01)和8倍(p<0.01)。100N处理的N2排放速率峰值(2226±77 μgN h–1 kg–1 d.s.)要比N2O和NO峰值滞后大约26 h,且分别比N2O和NO速率峰值高3倍(p<0.01)和5倍(p<0.01)。之后两处理的N2、N2O和NO排放速率均逐渐降低,10N处理3种气体依次在升温后107、30和30 h左右降到检测限附近,100N处 理则依次在升温后124、70和70 h左右降到检测限。此阶段两处理的CO2排放速率动态均呈现双峰型,第一次峰值出现时间均与N2O和NO一致,且峰值 排放速率分别为594和1060 μgC h–1 kg–1d.s.,第二次峰值排放速率分别为850和1368 μgC h–1 kg–1d.s.,出现时间比N2O和NO分别滞后28 h(10N)和24 h(100N)。100N处理的CO2排放显著高于10N处理,反硝化微生物利用DOC作为呼吸的电子供体,当底物DOC充足时,高的反硝化速率对应更多的电子传递,消耗更多的DOC,产生的更多的CO2(Swerts et al., 1996a)。
3.2 不同培养阶段的N2、N2O、NO和CO2排放总量与比值
反硝化氮素气体排放总量定义为N2、N2O和NO累积排放量之和(即Nt-N=N2-N+N2O-N+ NO-N,均以纯N计算)。CO2-C/Nt-N摩尔比对于验证反硝化过程基本理论有指示意义(Swerts et al., 1996a)。有研究结果表明,当微生物利用葡萄糖作为电子供体和能源时,反硝化产物以NO-N、N2O-N或N2-N为主时,CO2-C/Nt-N排放摩尔比分别为0.75、1.0和1.25(Reddy and DeLaune 2008;Swerts et al., 1996a,1996b)。当CO2-C/Nt-N摩尔比大于1.25时,认为CO2来自于其他过程,非反硝化过 程,并不伴随着含氮气体的排放;当CO2-C/Nt-N摩尔比小于0.75时,认为反硝化微生物利用其他的碳源,如丁酸,而非葡萄糖作为电子传递的供体和能量(Swerts et al., 1996a)。
在P1阶段,两处理的N2、N2O和NO累积排放总量均较低,可证明低温有氧的气体置换过程可很好的抑制微生物硝化及反硝化过程,保存底物氮。该阶段有氧,我们关注的是反硝化过程,所以不对该阶段的气体排放摩尔比进行讨论。
在P2阶段,两处理的N2、N2O、NO和CO2累积排放总量均呈极显著差异(p<0.001),100N比10N依次分别高2.2、3.8、2.5和1.5倍(表 2)。10N和100N的NO/N2O摩尔比分别约为2.0和1.3,差异极显著(p<0.01);N2O/N2 摩尔比分别约为0.12和0.21,差异也极显著(p<0.01);CO2- C/Nt-N摩尔比分别约为5.0和3.1,两者差异显著(p<0.05)。
|
|
表2 培养阶段的全部反硝化氮素气体(Nt=N2+N2O+NO)、单种反硝化氮素气体(>N2、N2O<、NO)和CO2的累积排放量 Table 2 Cumulative emissions of all nitrogen gases(Nt=N2+N2O+NO),individual nitrogen gases(N2,N2O,NO) and CO2 in each incubation period |
在P3阶段,两处理的N2、N2O、NO排放总量差异与初始氮底物供应量差异相当,即100N的三种气体排放总量分别比10N高9.5、8.6和12.3倍(p<0.0001),但CO2 排放总量的差异却不显著(表 2)。10N和100N在此阶段的NO/N2O摩尔比分别约为1.11和1.55,N2O/N2摩尔比分别约为 0.20和0.19,两个处理间的差异都不显著。对于整个P3阶段,10N和100N的CO2-C/Nt-N摩尔比 分别约为7.9和1.1,两处理间差异极显著(p<0.01)。从图 3可见,在P3阶段的前期,反硝化氮素气体排放明显发生,我们将这一反应阶段称为 反硝化阶段;随后,反硝化氮素气体排放不再明显,但CO2 排放依然比较强,我们将这一反应阶段称 为非反硝化阶段(Swerts et al., 1996a)。尽管初始NO3-含量水平相差9倍之多,但如表 2所示,两个处理在反硝化阶段的CO2-C/Nt-N摩尔比却在数值上比较接近,10N约为1.6,100N约为0.8,不过两个处理间的差异仍然极显著(p<0.01)。在之后的非反硝化阶段,两个处理的CO2-C/Nt-N摩尔比分别大幅度增加到大约311和58,且两者差异显著(p<0.05)。
1 0 N和100 N两处理的反硝化气体排放均主要发生在P3的反硝化阶段,两处理在此阶段的全部反硝化氮素气体总量Nt分别占三个阶段总和的85%和92%。10N处理在整个培养过程中的CO2排放总量有16%和68%分别发生在反硝化阶段和其后的非反硝化阶段,100N处理CO2排放总量有60%和21%分别发生在反硝化阶段和非反硝化阶段,由此可见,两个处理的CO2排放总量在这两个反应阶段的分配比例呈相反趋势(图 3)。
就整个培养过程而言,10N和100N两处理的反硝化氮素气体均以N2 为主,累积排放量分别为10±1和88±2 mgN kg–1d.s.,各占氮素气体排放 总量的77%和75%,其中10N显著高于100N(p<0.05)。初始NO3-水平对N2O累积量占氮素气体排放总量比值的影响不显著,10N为15%,100N为14%。NO排放占氮素气体排放总量的比重是10N低100N高(p<0.05),分别为8%和11%。整个培养过程的CO2-C/Nt-N摩尔比在两个处理之间差异极显著(p<0.01),10N为7.9,100N为1.1(表 2)。
3.3 整个培养过程前后的碳、氮平衡
整个培养过程前后的碳平衡用培养结束时的土壤DOC、SMBC含量与CO2累积排放量之和与培养刚开始时的土壤DOC、SMBC含量之和的百分比来表征。类似地,整个培养过程前后的氮平衡用培养结束时的土壤NH4+-N、NO3--N、SMBN含量与氮素气体排放总量Nt之和与培养刚开始时的土壤NH4+-N、NO3--N、SMBN含量之和的百分比来表征。培养过程中,土壤C素回收率用CO2累积排放量与培养前后土壤碳含量(DOC与SMBC含量之和)变化量的百分比来表示;土壤N素回收率用氮素气体累积排放总量(Nt=N2+N2O+NO)与培养前后土壤NO3--N变化量的百分比。培养前后的土壤DOC、NH4+-N、NO3--N、SMBN、SMBC含量和整个培养过程中的CO2和氮素气体排放总量如表 3所示。其中初始的DOC和NO3--N含量均为土壤自身含量与加入量之和。
| 表 3 培养前后土壤碳和氮素的平衡状况 Table 3 Carbon and nitrogen balance before and after the incubation |
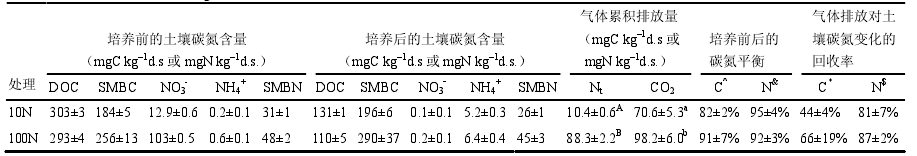
对于10N处理,培养后DOC含量极显著地下降到131 mgC kg–1d.s.(p<0.0001),SMBC含量有所增加(197 mgC kg–1d.s.),但不显著,SMBN和NO3--N分别减少到26 mgN kg–1d.s.(p=0.05)和0.1 mgN kg–1d.s.(p<0.0001),NH4+-N显著增加到5.2 mgN kg–1d.s.(p<0.001)。100N处理,培养后的DOC含量显著降低到111 mgC kg–1d.s.(p<0.0001),SMBC含量略有增加(290 mgC kg–1d.s.),SMBN略有减少(45 mgN kg–1d.s.),两者变化都不显著,NO3--N含量极显著地降低到0.2 mgN kg–1d.s.(p<0.0001),NH4+-N含量极显著地增加到6.4 mgN kg–1d.s.(p<0.001)。
从表 3可见,10N处理培养前后的碳、氮平衡分别为82%和95%,100N处理分别为91%和92%;10N和100N处理在整个培养过程中的CO2累积排放量测定值对培养前后土壤碳含量(DOC与SMBC含量之和)变化的回收率分别为44%和66%,氮素气体排放总量测定值对培养前后土壤NO3--N含量变化的回收率分别为81%和87%。
4 讨论 4.1 测定系统与方法的有效性
![]()
![]()
![]() 从以上结果可见,培养前后测定的土壤碳素、氮素的平衡结果较好(82%~95%);通过测定反硝化氮素气体排放可以回收培养前后的土壤NO3–-N含量变化81%~87%;整个培养过程中的土壤反硝化氮素气体和CO2排放速率动态可很好地验证反硝化过程基本理论。这三点可说明该氦环境培养—气体同步直接测定法能够比较准确地定量土壤反硝化气体排放速率及其产物比。
从以上结果可见,培养前后测定的土壤碳素、氮素的平衡结果较好(82%~95%);通过测定反硝化氮素气体排放可以回收培养前后的土壤NO3–-N含量变化81%~87%;整个培养过程中的土壤反硝化氮素气体和CO2排放速率动态可很好地验证反硝化过程基本理论。这三点可说明该氦环境培养—气体同步直接测定法能够比较准确地定量土壤反硝化气体排放速率及其产物比。
该测定系统和方法在整个培养过程中检测的氮素气体(N2、N2O和NO)和CO2排放速率动态与反硝化过程基本理论比较吻合。
低温抑制微生物活性,有氧抑制反硝化作用(Knowles,1982),这从P1阶段测得的非常低的N2O、NO和CO2排放速率得到了很好的体现。在这一阶段,初始碳底物(DOC)被气体排放消耗1%~3%,而初始氮底物(NO3--N)的消耗率不到1/1000,这表明完全达到了设计低温有O2置换条件的目的,即有效阻止置换过程中呼吸作用、硝化作用等氧化过程及反硝化作用造成底物消耗。
为了测定反硝化过程,必须将土壤从有氧培养环境转换为无氧培养环境,因而,测定系统设计了P2阶段,在P2阶段,土壤空气中的O2含量迅速减少(完全清除O2大约需5 h左右),反硝化作用增强,使测得的N2、N2O和NO排放速率较上阶段明显增加,但由于受温度(2 ℃)的限制,这些气体的排放速率较低。这一阶段的气体排放导致了2%~4%的初始碳底物和4%(100N)~12%(10N)的初始氮底物被消耗。该氮消耗率表明,控制这一阶段反硝化作用强度的不是底物有效性,而是温度。
如果培养土壤处于低初始N水平和高初始DOC-C/NO3--N质量比条件下(例如10N,初始N水平为13.6 mgN kg–1d.s.,DOC-C/NO3--N质量比为22),则在P3的反硝化阶段,三种氮素气体排放速率峰值几乎同时出现,此时也出现第一个CO2排放速率峰值,第二个CO2排放速率峰值明显滞后于N2约26 h,而且发生在非反硝化阶段(图 3a),这与Swerts et al.(1996a)的研究结果相一致,造成两者一致的原因可能跟土壤初始DOC-C/NO3--N质量比较一致有关[本研究10N处理的土壤初始DOC-C/NO3--N质量比约为22,Swerts et al.(1996a)的约为10]。10N处理在反硝化阶段的CO2-C/Nt-N摩尔比为1.6,CO2-C/Nt-N摩尔比大于1.25表明培养过程中排放的CO2可能来源于其他微生物过程(并不伴随着含N气体的排放),如微生物参与的Fe3+和SO42–还原(Yao et al., 1999),或者厌氧发酵过程(Swerts et al., 1996a),或者厌氧矿化过程(Bridgham et al., 1998)。其他微生物过程贡献CO2排放可从10N处理含N气体和CO2排放动态中得到证实,即CO2排放峰再次出现而此时含N气体的排放已接近系统检测线了。随着土壤中NO3-在反硝化过程中被耗尽,培养土壤进入非反硝化阶段,这时,剩余的DOC及死亡微生物的发酵反应促成了第二个CO2排放峰。之后,随着土壤中的DOC逐渐被消耗,发酵作用逐渐减弱,CO2排放也随之逐渐减少,并最终稳定在一定水平(图 3a),这个稳定水平在很大程度上可能由微生物维持呼吸强度所决定。
如果培养土壤处于高初始N水平和低初始DOC-C/NO3--N质量比条件下(例如100N,初始 N水平为103 mgN kg–1d.s.,DOC-C/NO3--N质量比为2.8),则在P3的反硝化阶段,N2O与NO排放峰值和第一个CO2排放峰值同时出现,N2 排放峰值明显滞后,但却与第二个CO2排放峰值同步(图 3b)。在100N处理的N2O和NO排放达到峰值之前,反硝化中间产物与最终产物的质量比N2O/N2和(N2O+NO)/N2分别为1.2和1.9。这证实了有关反硝化理论阐述:在NO3--N供应比较充足的 情况下,反硝化会优先利用NO3--N作为电子受体,将其还原为N2O和NO等中间产物,从而造成N2O和NO的积累(Nömmik,1956;Swerts et al., 1996c)。此反硝化阶段的CO2-C/Nt-N摩尔比为0.8,与反硝化反应的理论比值范围0.80~1.25的下限比较一致。随着NO3-逐渐被耗尽,N2O和NO代替NO3-成为反硝化的电子受体,并最终被还原为N2,从而形成N2 排放峰,与此同时,反硝化过程产生的CO2 也导致其第二个排放峰值。之后,随着氮底物被逐渐耗尽,反硝化作用逐渐停止,氮素气体排放速率降低到检测限附近。
4.3 培养前后的氮平衡
本研究的测试结果显示,无论初始NO3--N含量处于低水平还是高水平,反硝化气体累积排放量可回收培养前后土壤NO3--N库变化的81%~87%,N素回收率计算涉及4个变量,考虑每一个变量均存在测定误差,该回收率结果比较令人满意。反硝化过程的消耗使培养结束时的NO3--N含量降低到检测限附近,而NH4+-N含量培养结束后则由最初的<1 mgN kg–1d.s.增加到6 mgN kg–1d.s.左右。培养过程中NH4+-N含量增加可能有四条途径:一是NO3-异化还原为NH4+(DNRA)。有资料证明,有的细菌在无氧条件下能使NO3-异化还原为NH4+(Caskey and Tiedje, 1979;McCready et al., 1983;Tiedje,1988;李振高等,1989);二是厌氧条件下的土壤有机氮矿化(Bridgham et al., 1998)。旱地土壤中存在大量的硫酸盐和氧化态的铁和锰,当土壤空气中的O2被置换清除后,这些氧化态的物质有可能成为有机质降解的主要电子受体,使其厌氧矿化产生NH4+;三是死亡微生物细胞溶解释放;四是厌氧条件下的非共生固氮(Rice and Paul, 1972;Swerts et al., 1996a,1996b)。
此外,100N处理中的SMBN变化量比NH4+-N增加量少,也不能排除是测定误差所致。本研究给出SMBN含量时,没有对SMBN测定结果进行提取率校正,因为不同土壤的提取率通常存在差异,而目前我们尚不能确定测定土壤的准确提取率。由此可能使给出的数据比实际值偏小,SMBN含量越高,偏差量可能越大。
4.4 反硝化产物摩尔比[NO/N2O、NO/(N2O+N2)和N2O/N2]
NO/N2O摩尔比是否大于1,一直作为土壤的排放NO和N2O来自于硝化作用还是反硝化作用的判据。一般认为,如果NO/N2O摩尔比<1,是反硝化作用占主导(Anderson and Levine 1986)。本研究中,10N和100N两处理在整个培养过程中反硝化中间产物排放总量的NO/N2O摩尔比分别为1.2和1.5,两者无显著差异,但却都显著高于1.0(p<0.001),这与以往对于反硝化中间产物比的认识不一致。基于厌氧条件下液体培养基培养反硝化细菌和硝化细菌的试验结果,Anderson and Levine(1986)发现,硝化细菌作用下形成的NO/N2O摩尔比大于1,而反硝化细菌作用下形成的NO/N2O摩尔比却远小于1。本研究结果表明,对于置于氦环境中培养的水分含量约为53% WFPS的土壤,其反硝化产物的NO/N2O摩尔比要高于水分饱和的基质(如液体培养基)中的反硝化NO/N2O摩尔比。导致如此差异的原因可能是,在水分不饱和情况下(如本研究培养土壤的情形),反硝化产生的NO扩散离开培养基质的过程要比水分饱和条件下(如液体培养基和淹水的情形)快得多,因而被反硝化细菌捕捉并进一步还原为N2O(Robertson and Groffman, 2007)的机会就会小得多,从而导致高于1.0的NO/N2O摩尔比。这意味着,不能用NO/N2O摩尔比大于1与否来作为土壤排放的N2O和NO是主要来源于硝化作用还是反硝化作用的判定依据。
在无O2条件下采用乙炔抑制法测定水分不饱和土壤的反硝化速率时,所测得的N2O排放量实际上是反硝化排放的N2O和N2之和。本研究中,10N和100N两处理在整个培养过程中排放气体的NO/(N2O + N2)质量比分别为0.09和0.12,后者显著高于前者(p<0.05)。这意味着,如果像绝大多数研究一样不同时检测NO排放,则无O2条件下水分不饱和土壤的反硝化速率可能会被乙炔抑制法低估9%~12%,低估程度因土壤初始NO3–含量 多少而异。
对N2O/N2摩尔比的测定是估算土壤N2排放的重要指标(Groffman et al., 2006)。有研究指出,反硝化产物的N2O/N2摩尔比随土壤初始NO3–含量的增加而增大(Focht,1985;Sahrawat and Keeney, 1986;范晓晖和朱兆良,2002)。反硝化的N2O/N2比大小很大程度上取决于氧化剂(NO3-)和还原剂(有效碳)的相对有效性。当氧化剂NO3-供应超过反硝化对还原剂有效碳的需求时,反硝化就会因缺少电子而进行不彻底,生成较多中间产物NO和N2O,从而导致N2O/N2比增加;反之,反硝化过程则进行得很彻底,生成较多的终产物N2,导致N2O/N2比下降(Hutchinson and Davidson, 1993)。本研究中10N和100N两处理的N2O/N2摩尔比均为0.19,两者无差异。尽管100N处理的NO3-含量显著高于10N处理,但两处理的DOC-C/NO3—N 的摩尔比均>1.25,反硝化过程的底物碳供应充足,反硝化过程进行彻底,产物以N2为主。除底物NO3-和DOC外,其他环境因子(如土壤氧气状况、温度和湿度、pH值、质地等)也会影响反硝化产物的N2O/N2比(Weier et al., 1993;Dannenmann et al., 2008;Cuhel et al., 2010),导致不同情况下的N2O/N2比值差异较大,如Scheer et al.(2009)观测到的N2O/N2比值可在0.005~0.2之间变化。
5 结论本研究的测试结果表明:通过测定反硝化氮素气体——氮气、氧化亚氮和一氧化氮的排放速率动态,Wang et al.(2011)新建的氦环境培养-气体同步直接测定系统和方法回收培养土壤中消失的NO3–的81%~87%;结合土壤无机氮和微生物氮含量测定,该方法可使培养前后的氮平衡率达92%~95%;在氮底物比较充分时,反硝化阶段排放的二氧化碳与反硝化氮素气体总量的摩尔比与反硝化反应的理论值具有较好的一致性。这些充分显示,该新建系统和测定方法对土壤反硝化过程的测定具有很好的可靠性,为定量研究土壤反硝化过程提供了有效的直接测定手段。
致谢 对本研究提供技术支持和帮助的有中国科学院大气物理研究所的刘广仁、张文、王迎红、廖婷婷、李宝江、罗献宝等,谨此致谢!
| [1] | Anderson I C, Levine J S. 1986. Relative rates of nitric oxide and nitrous oxide production by nitrifiers, denitrifiers, and nitrate respirers[J]. Applied and Environmental Microbiology, 51 (5): 938-945. |
| [2] | 鲍士旦. 2000. 土壤农化分析[M]. 北京: 中国农业出版社, 49-56. Bao Shidan. 2000. Soil Agricultural Chemistry Analysis (in Chinese)[M]. Beijing: China Agriculture Press, 49-56. |
| [3] | Bollmann A, Conrad R. 1997. Acetylene blockage technique leads to underestimation of denitrification rates in oxic soils due to scavenging of intermediate nitric oxide[J]. Soil Biology and Biochemistry, 29 (7): 1067-1077. |
| [4] | Bridgham S D, Updegraff K, Pastor J. 1998. Carbon, nitrogen, and phosphorus mineralization in northern wetlands[J]. Ecology, 79 (5): 1545-1561. |
| [5] | Butterbach-Bahl K, Willibald G, Papen H. 2002. Soil core method for direct simultaneous determination of N2 and N2O emissions from forest soils[J]. Plant and Soil, 240 (1): 105-116. |
| [6] | Cárdenas L M, Hawkins J M B, Chadwick D, et al. 2003. Biogenic gas emissions from soils measured using a new automated laboratory incubation system[J]. Soil Biology and Biochemistry, 35 (6): 867-870. |
| [7] | Caskey M H, Tiedje J M. 1979. Evidence for clostridia as agents of dissimilatory reduction of NO3- to H4+ in soils[J]. Soil Science Society of America Journal, 43 (5): 931-936. |
| [8] | Cuhel J, Simek M, Laughlin R J, et al. 2010. Insights into the effect of soil pH on N2O and N2 emissions and denitrifier community size and activity[J]. Applied and Environmental Microbiology, 76 (6): 1870-1878. |
| [9] | Dannenmann M, Butterbach-Bahl K, Gasche R, et al. 2008. Dinitrogen emissions and the N2: N2O emission ratio of a Rendzic Leptosol as influenced by pH and forest thinning[J]. Soil Biology and Biochemistry, 40 (9): 2317-2323. |
| [10] | Davidson E A, Seitzinger S. 2006. The enigma of progress in denitrification research[J]. Ecological Applications, 16 (6): 2057-2063. |
| [11] | 范晓晖, 朱兆良. 2002. 旱地土壤中的硝化—反硝化作用[J]. 土壤通报, 33 (5): 385-391. Fan Xiaohui, Zhu Zhaoliang. 2002. Nitrification and denitrification in upland soils[J]. Chinese Journal of Soil Science (in Chinese), 33 (5): 385-391. |
| [12] | Focht D D. 1985. Differences in nitrogen-15 enrichments of evolvednitrous oxide and dinitrogen and the question of auniform nitrate-15 pool[J]. Soil Science Society of America Journal, 49 (3): 786-790. |
| [13] | Groffman P M, Altabet M A, Bohlke J K, et al. 2006. Methods for measuring denitrification: diverse approaches to a difficult problem[J]. Ecological Applications, 16 (6): 2091-2122. |
| [14] | Hutchinson G L, Davidson E A. 1993. Processes for production and consumption of gaseous nitrogen oxides in soil[M].//Harper L A, Mosier A R, Duxbury J M, et al. Agricultural Ecosystem Effects on Trace Gases and Global Climate Change. Madison, WI: ASA Special Publication, 79-93. |
| [15] | IPCC. 2007. Climate Change 2007: The Physical Scientific Basis[M].//Contribution of Working Group 1 to the Fourth Assessment Report of the Intergovernmental Panel on Climate Chang. Solomon S, Qin D, Manning M, et al., Eds. Cambridge, United Kingdom and New York, NY, USA: Cambridge University Press, 996. |
| [16] | Jordan T E, Weller D E, Correll D L. 1998. Denitrification in surface soils of a riparian forest: Effects of water, nitrate, and sucrose[J]. Soil Biology and Biochemistry, 30 (7): 833-843. |
| [17] | Klemedtsson L, Hansson G, Mosier A. 1990. The use of acetylene for the quantification of N2 production from biological processes in soil[M].//Revsbech J P, Sorensen J. Denitrification in Soil and Sediment. New York: Plenum Press, 167-180. |
| [18] | Knowles R. 1982. Denitrification[J]. Microbiological Reviews, 46 (1): 43-70. |
| [19] | 李明, 梁旺国, 郑循华, 等. 2009. 晋南地区典型盐碱地棉田的NO排放特征[J]. 气候与环境研究, 14 (3): 318-328. Li Ming, Liang Wangguo, Zheng Xunhua, et al. 2009. Characteristics of NO emission from typical saline soil of southern Shanxi cotton land[J]. Climatic and Environmental Research (in Chinese), 14 (3): 318-328. |
| [20] | 李振高, 潘映华, 伍期途, 等. 1989. 太湖地区水稻土优势反硝化细菌的数量、组成与酶活性[J]. 土壤学报, 26 (1): 79-86. Li Zhengao, Pan Yinghua, Wu Qitu, et al. 1989. Numbers, compositions and enzyme activities of denitrifiers in paddy soils of Taihu Lake district[J]. Acta Pedologica Sinica (in Chinese), 26 (1): 79-86. |
| [21] | McCready R G L, Gould W D, Brendregt R W. 1983. Nitrogen isotope fractionation during the reduction of NO3-to NH4+ by Desulfovibrio sp.[J]. Canadian Journal of Microbiology, 29 (2): 231-234. |
| [22] | Molstad L, Dörsch P, Bakken L R. 2007. Robotized incubation system for monitoring gases (O2, NO, N2O, N2) in denitrifying cultures[J]. Journal Microbiological Methods, 71 (3): 202-211. |
| [23] | Nömmik H. 1956. Investigations on denitrification in soil[J]. Acta Agriculturae Scandinavica, 6 (2): 195-228. |
| [24] | Reddy K R, DeLaune R D. 2008. Biogeochemistry of Wetlands: Science and Applications[M]. CRC Press: Boca Raton: 136-151. |
| [25] | Rice W A, Paul E A. 1972. The organisms and biological processes involved in asymbiotic nitrogen fixation in waterlogged soil amended with straw[J]. Canadian Journal of Microbiology, 18 (6): 715-723. |
| [26] | Robertson G P, Groffman P M. 2007. Nitrogen Transformations[M].//Paul E A. Soil Microbiology, Ecology, and Biochemistry. 3rd ed. Amsterdam, Boston, Heidelberg, London, New York, Oxford, Paris, San Diego, San Francisco, Singapore, Sydney, Tokyo: Elsevier, 341-364. |
| [27] | Sahrawat K L, Keeney D R. 1986. Nitrous oxide emission from soils[J]. Advance in Soil Science, 4: 103-148. |
| [28] | Scheer C, Wassmann R, Butterbach-Bahl K, et al. 2009. The relationship between N2O, NO, and N2 fluxes from fertilized and irrigated dryland soils of the Aral Sea Basin, Uzbekistan[J]. Plant Soil, 314 (1-2): 273-283. |
| [29] | Scholefield D, Hawkins J M B, Jackson S M. 1997a. Development of a Helium atmosphere soil incubation technique for direct measurement of nitrous oxide and dinitrogen fluxes during denitrification[J]. Soil Biology and Biochemistry, 29 (9-10): 1345-1352. |
| [30] | Scholefield D, Hawkins J M B, Jackson S M. 1997b. Use of a flowing helium atmosphere incubation technique to measure the effects of denitrification controls applied to intact cores of a clay soil[J]. Soil Biology and Biochemistry, 29 (9-10): 1337-1344. |
| [31] | Simarmata T, Benkiser G, Ottow J C G. 1993. Effect of an increasing carbon: nitrate-N ratio on the reliability of acetylene in blocking the N2O-reductase activity of denitrifying bacteria in soil[J]. Biology and Fertility of Soils, 15 (2): 107-112. |
| [32] | Stefanson R C, Greenlan D J. 1970. Measurement of nitrogen and nitrous oxide evolution from soil-plant systems using sealed growth chambers[J]. Soil Science, 109 (3): 203-206. |
| [33] | Swerts M, Uytterhoeven G, Merckx R, et al. 1995. Semicontinuous measurement of soil atmosphere gases with gas-flow soil core method[J]. Soil Science Society of America Journal, 59 (5): 1336-1342. |
| [34] | Swerts M, Merckx R, Vlassak K. 1996a. Denitrification, N2-fixation and fermentation during anaerobic incubation of soils amended with glucose and nitrate[J]. Biology and Fertility of Soils, 23 (3): 229-235. |
| [35] | Swerts M, Merckx R, Vlassak K. 1996b. Denitrification followed by N2 fixation during anaerobic incubation[J]. Soil Biology and Biochemistry, 28 (1): 127-129. |
| [36] | Swerts M, Merckx R, Vlassak K. 1996c. Influence of carbon availability on the production of NO, N2O, N2, CO2 by soil cores during anaerobic incubation[J]. Plant and Soil, 181 (1): 145-151. |
| [37] | Sparling G P, West A W. 1988. Modifications to the fumigation-extraction technique to permit simultaneous extraction and estimation of soil microbial-C and microbial-N[J]. Communications in Soil Science and Plant Analysis, 19 (3): 327-344. |
| [38] | Tiedje J M. 1988. Ecology of denitrification and dissimilatory nitrate reduction to ammonium[M]//Zehnder A J B. Biology of Anaerobic Microorganisms. New York: Wiley. |
| [39] | Wang R, Willibald G, Feng Q, et al. 2011. Measurements of N2, N2O, NO and CO2 emissions from soils with the gas-flow-soil-core technique[J]. Environmental Science and Technology, 45 (14): 6066-6072. |
| [40] | Weier K L, Doran J W, Power J F, et al. 1993. Denitrification and the dinitrogen/nitrous oxide ratio as affected by soil water, available carbon, and nitrate[J]. Soil Science Society of America Journal, 57 (1): 66-72. |
| [41] | Yao H, Conrad R, Wassmann R, et al. 1999. Effect of soil characteristics on sequential reduction and methane production in sixteen rice paddy soils from China, the Philippines, and Italy[J]. Biogeochemistry, 47 (3): 269-295. |
| [42] | Yoshinari T, Knowles R. 1976. Acetylene inhibition of nitrous oxide reduction by denitrifying bacteria[J]. Biochemical and Biophysical Research Communications, 69 (3): 705-710. |
| [43] | Zheng X H, Mei B L, Wang Y H, et al. 2008. Quantification of N2O fluxes from soil-plant systems may be biased by the applied gas chromatograph methodology[J]. Plant and Soil, 311 (1-2): 211-234. |